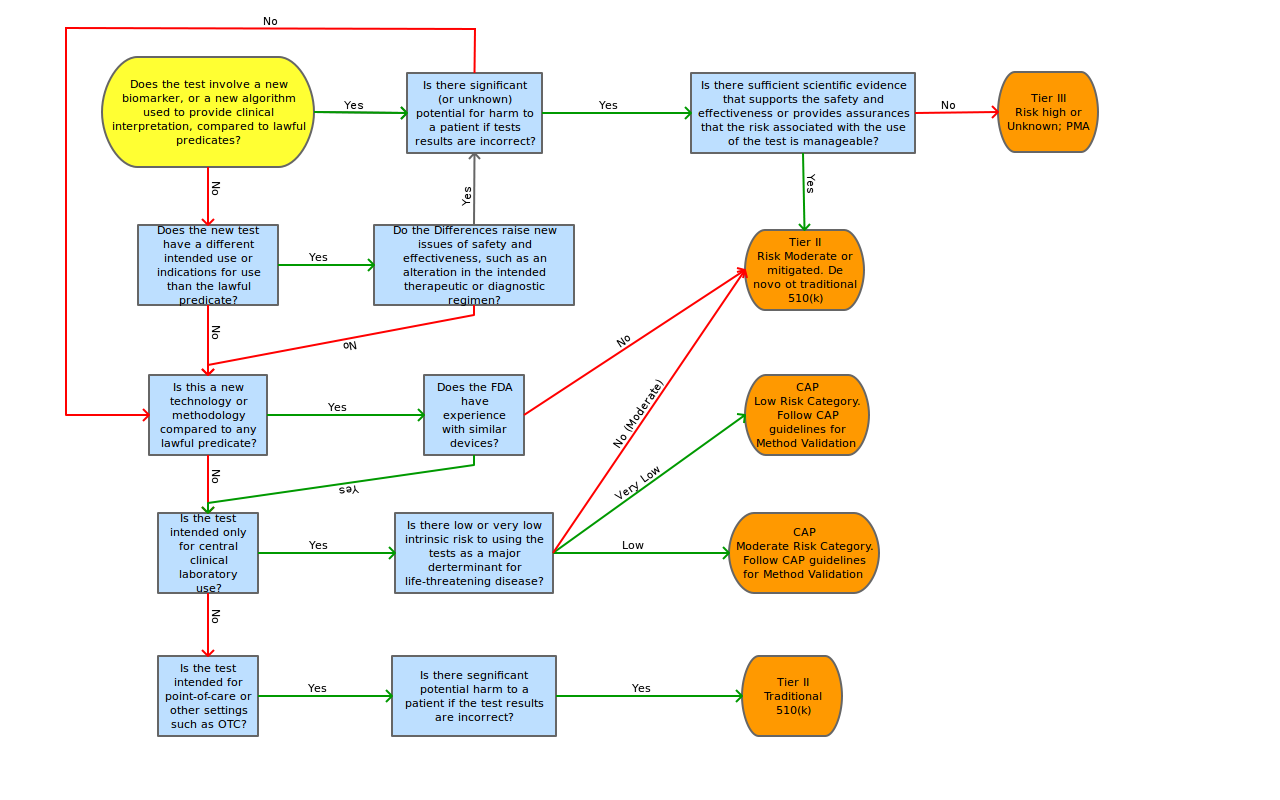
Introduction
The completion of Human Genome Project marks a defining moment in the history of medical science. New technologies in molecular diagnostics and computational biology have furthered understanding of the transcriptome and proteome at a rate previously unimaginable. Since then, a flood of gene expression profile tests and other biomarker panels have been introduced to the clinical diagnostics market. These tests were all developed in a similar manner—by measuring and comparing the abundance of a group of specific gene transcripts, proteins, or other biomarkers in populations of patients or tissues with a known pathology or prognosis state. This comparative analysis results in the development of a predictive algorithm that can determine the probability of disease or prognosis in unknown samples.
On July 26 2007, the Food and Drug Administration (FDA) published a draft guidance to address this emerging field of In Vitro Diagnostics for Multivariate Index Assays (IVDMA). An IVDMIA is defined by the FDA as a device that:
- Combines the values of multiple variables using an interpretation function to yield a single, patient-specific result (e.g., a “classification,” “score,” “index,” etc.), that is intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment or prevention of disease, and
- Provides a result whose derivation is non-transparent and cannot be independently derived or verified by the end user.As a result, several organizations filed their products for de novo 510(k) clearance, while most waited for a final guidance. The topic has since been shelved by the FDA as they now consider broadening the guidance to govern complex Laboratory Developed Tests (LDTs) currently governed under CLIA. Although the final guidance has been postponed, the final version will most likely still contain most of the items identified in the draft.
Purpose
The purpose of this document is to summarize the draft guidance as it pertains to Laboratory Developed Tests and discuss its implications at an independent clinical laboratory.
Scope
The scope of this document is limited to studying the draft guidance as it was written in 2007. It does not contain any commentary regarding any potential changes to the guidance as a response to industry comments or a changing regulatory climate.
Analysis
Approach
The FDA continues its efforts at administrating a “least burdensome approach” for IVDMIA. The guidance states that, although there is a potential for complicated and emerging technologies to be included in IVDMIA submissions, the classification of devices will be based on risks associated with intended use. (The guidance includes an example of a test that indicates the likely prognosis of a cancer will most likely be considered Class II, versus a test that indicates the therapy regimen would be considered Class III.) The FDA acknowledges the fact the nature of the types of disease states analyzed by these new tests will most likely require a classification of II or III.
The guidance states that safety and effectiveness determination should include “review of the performance of the entire system, including the accurate measurement of the input variables, directions for use, and expected analytical or clinical performance, rather than a review of only certain subcomponents of the test” (i.e. just the algorithm). This approach is consistent with classification determination of other devices, like clinical chemistry and clinical toxicology test systems.
Premarket Regulation
In the spirit of using a “least burdensome approach”, the FDA proposes a flexible approach to safety and effectiveness determination. Although a prospective study is preferred, the administration will also consider archived samples and/or retrospective studies, as long as the study design, sample composition, sample selection and sample storage processes reflect the intended use and intended population for use of the device.
In lieu of properly designed retrospective studies, IVDMIA manufacturers may also file for “Investigational Use Only” labeling as defined by 21 CFR 809.10 and apply for an investigational device exemption (IDE). Other labeling requirements are to be consistent with those regarding other devices.
Postmarket Regulation
Consistent with other devices, the FDA states that IVDMIAs are subject to Quality System regulation as described in 21 CFR Part 820, however, the FDA intends to continue to exercise enforcement discretion for CLIA-regulated laboratories until a final guidance is approved for laboratory developed tests.
The Administration states that all IVDIAs must comply with 21 CFR 803 – Medical Device
Reporting standards. Also, laboratories that run IVDMIA test systems are considered “user facilities”, and must submit serious injury/device malfunction reports as such (CFR 21 830.50).
Enforcement
In an effort to reduce the effect on innovation costs in the market, FDA intends to exercise enforcement discretion with respect to all regulatory requirements for currently marketed, laboratory-developed IVDMIAs for 12 months following publication of the final guidance document. In an effort to encourage early adoption to the new standards, FDA intends to exercise enforcement discretion for an additional 6 months for any currently marketed, laboratory-developed IVDMIAs if the manufacturer submits a 510(k) or PMA within the initial 12 month period following publication of the final guidance.
Conclusion
While the debate on whether Laboratory Developed Tests are medical services (and should be regulated by CMS under CLIA) or medical devices (and should be regulated by the FDA as IVDs) seems to be coming to a head, LDT developers and IVD manufactures brace for a dramatic shift in regulatory environment. FDA has used “enforcement discretion” in exerting what it sees as its legal authority to regulate medical devices, but many analysts believe that the time will soon come when enforcement will be applied to some areas currently left up to CLIA. In particular, multivariate index assays have come under particular attention in recent years; enough to spur publication of draft guidance by the FDA.
This emerging development may provide some interesting opportunites for device manufacturers. Since they many systems already in place to govern compliance to FDA device manufacturing requirements for new and existing products, these systems can and should be leveraged in order to bring their own CLIA laboratory into compliance for a potential 510(k) or PMA submission when/if the time comes. Other leaders in IVDMIA laboratory testing have already begun to retrofit their current processes and build their new products with the FDA in mind, but without the institutional regulatory architecture, they will be at a disadvantage. Life Technologies has the opportunity to create IVDMIA products with competitive compliance that surpasses the expectations of regulators and their customers while controlling costs.
References
- 2007. Food and Drug administration. Draft Guidance for Industry, Clinical Laboratories, and FDA Staff. In Vitro Diagnostic Multivariate Index Assays. http://www.fda.gov
- 2011. Smith, Katie M. Exploring FDA-Approved IVDMIAs. http://www.ivdtechnology.com/article/exploring-fda-approved-ivdmias
- 2012. Wiess, Ronald L. The Long and Winding Regulatory Road for Laboratory-Developed Tests. American Journal of Clinical Pathology, 138, 20-26.
NO EVENT SHALL LAB INSIGHTS, LLC BE LIABLE, WHETHER IN CONTRACT, TORT, WARRANT, OR UNDER ANY STATUTE OR ON ANY OTHER BASIS FOR SPECIAL, INCIDENTAL, INDIRECT, PUNITIVE, MULTIPLE OR CONSEQUENTIAL DAMAGES IN CONNECTION OR ARISING FROM LAB INSIGHTS, LLC SERVICES OR USE OF THIS DOCUMENT.